




In science, working collaboratively is absolutely essential to increase the odds for success. Collectively pooling ideas can lead to better solutions, faster. If we want to achieve more, we need to join efforts and work smarter, not just harder. This is especially true in modern digital chemistry, where innovators might be searching for one single molecule among a nearly endless universe of possible candidates.
Working together to explore the vastness of chemical space requires a shared platform specifically designed to offer targeted solutions for specific workflows. We’re living in a digital revolution where high-speed computational tools are key to success. Advanced computations now make it possible to iterate through broad chemical space and predict how a molecule might behave in a given environment with a high degree of accuracy.
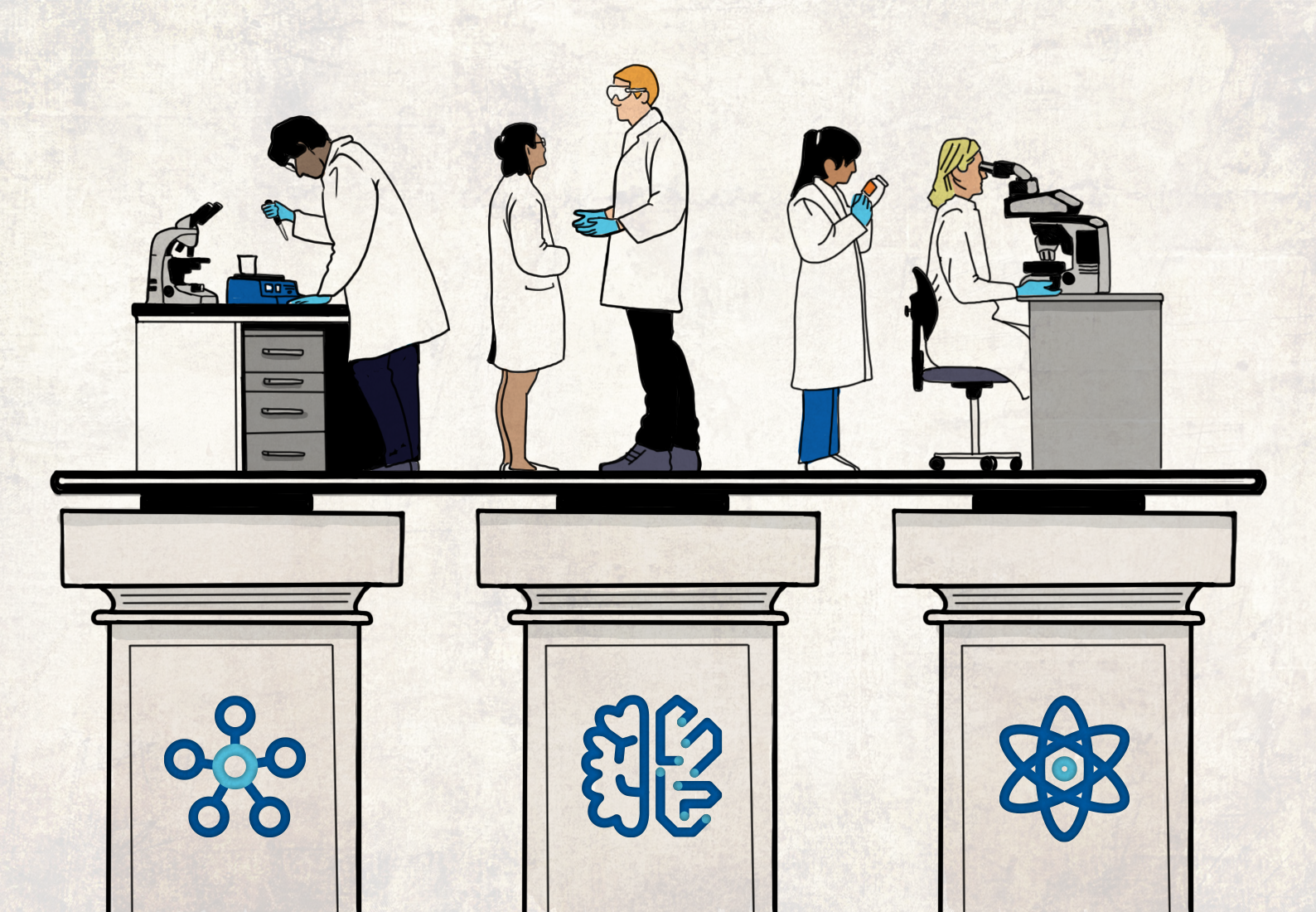
For all these reasons, chemistry innovations are taking place not only in the lab but more in the mind, through the cloud, and via computer simulations. We must embrace new ways of working in an increasingly digitized and collaborative world. As we search for new chemistry breakthroughs, I see three synergistic pillars at work.
By utilizing all three pillars in an integrated way, you’re no longer just managing your project, you’re elevating it.
 Pillar 1: Physics-based modeling
Pillar 1: Physics-based modeling
Each year, some of the most important discoveries involve molecules that serve specific human needs, like medicines or materials. To identify those molecules, scientists use their knowledge and experience designing compounds with desired physico-chemical properties. Researchers must consider how atoms behave when they bond to each other, form new molecules, and then interact with their environment—be it other molecules, solvents, or as part of a larger system.
But, again, one scientist working alone can only go so far.
That’s where physics-based modeling provides a real boost. It allows accurate prediction of a molecule’s properties, even in cases where properties and predictions might look counterintuitive based on general knowledge. The list of molecular properties that are amenable to accurate assessment using physics-based methods continues to grow rapidly and includes, but is not limited to, our ability to predict potency, selectivity, permeability, solubility and other parameters. This is, in effect, true digital chemistry.
However, rigorous physics-based computational methods are still rather expensive and require expertise to utilize. On a practical level, they generally can’t be cost- and time-effective when applied alone on an ultra-large scale.
Successful digital chemistry is not about the technology on its own. It’s about passionate, creative teams of scientists who embrace a technology-first culture.
 Pillar 2: Machine learning
Pillar 2: Machine learning
It’s now possible to layer the additional strengths of machine learning to amplify the impact of physics-based modeling. The speed of machine learning models that are trained on high quality datasets derived from physics-based modeling allows us to assess vast chemical space. A small subset of top ranking compounds emerging from these rapid predictive calculations can then be evaluated using highly accurate physics-based methods to confirm predictions and generate additional data for training the next generation of the machine learning model. This iterative approach provides a unique opportunity for researchers to evaluate billions of molecules computationally in a fraction of the time.
Importantly, many critical parameters in drug design can be evaluated in silico using these iterative approaches prior to synthesis, allowing for what is known as multi-parameter optimization (MPO). We often don’t have to make individual molecules to test each of the hypotheses generated computationally, but rather synthesize very few compounds that meet multiple requirements through simulations. High quality molecules therefore emerge early in the process due to the ability to optimize them in silico, and thus only the really promising ones are actually synthesized and tested. The learnings from each round of experimental testing refine and steer the next round of design and computational multi-parameter optimization. In this way, we’re experimenting a lot more in a high throughput fashion on the computer rather than doing it experimentally in the lab. The end result is that the best molecules are made sooner in the process, with a higher chance of success.
 Pillar 3: Effective enterprise informatics platform
Pillar 3: Effective enterprise informatics platform
Successful digital chemistry is not about the technology on its own. It’s about passionate, creative teams of scientists who embrace a technology-first culture. That leads us to the third pillar, where true collaboration drives success—not just between scientists, but between an individual scientist and the technology.
It starts with the power of informatics, which is using diverse types of data to solve complex problems. These data are often shared among multiple sources. For example, in healthcare, an informatics platform might include teaching hospitals, researchers, clinicians, electronic patient records, and DNA sequencing. With informatics, big data can be stored, accessed, searched, analyzed, and mined for new discoveries and connections.
Schrödinger’s approach to enterprise informatics is about empowering the scientist to have easy access to not just all of their experimental and predictive data, but the ability to design new molecules and immediately test them using physics-based modeling and machine learning. This brings the iterative design-model-make-test-analyze (DMMTA) cycle into the hands of the scientists, visualizing their data in whatever way they prefer. Being able to programmatically access and understand data is a natural opening to building machine learning models alongside these digital chemistry models, effectively delivering the amplification technology of machine-learning on top of a rich program management technology.
For disseminated teams in different cities and time zones, a platform geared at collaboration and live data is essential: always ready, always evolving.
By utilizing all three pillars in an integrated way, you’re no longer just managing your project, you’re elevating it. The collaborative process accelerates design cycles and generates more ideas. This approach leads the way to high-quality preclinical and clinical candidates and materials, in less time and at lower cost than traditional methods.
It’s an exciting time to be in digital chemistry, where partnering with people and collaborating in silico are the new ways to find the future.