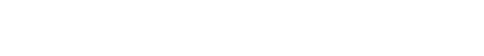Drug hunters face enormous challenges in their efforts to bring new treatments to patients. The traditional process of designing molecules that have the right properties to become safe and effective drug candidates takes approximately five years, requires thousands of molecules to be synthesized in the laboratory for preclinical testing, and costs many millions of dollars. And after all that effort, more than two-thirds of drug discovery programs fail to even deliver a small molecule development candidate suitable to take forward into clinical testing.
The long timeline, hefty price tag and high failure rate is due to the fact that many of the most important molecular properties, such as potency, selectivity, and solubility, are anti-correlated, meaning that it is difficult to design for one drug property (eg, potency) without negatively impacting another desirable property (eg, solubility).
It’s a “whack-a-mole” problem.
In an ideal world, we would be able to list out every possible molecule on a computer, feed these into an algorithm that computes every single molecular property with perfect accuracy, and then choose the best molecule to advance into the clinic. This is our North Star.
Predicting Molecular Properties
There are two distinct approaches being used within drug discovery that are making progress predicting an increasing number of molecular properties: artificial intelligence (AI)/machine learning and physics-based methods.

AI/Machine Learning
AI is a broad umbrella term simply meaning machines, particularly computers, that perform tasks typically conducted by humans, such as autonomously driving a car and identifying faces in a crowd. The branch of AI involved in drug discovery and development is called machine learning. Machine learning systems are trained to comb through large amounts of existing data, called a training set, to detect patterns, predict outcomes, and make decisions with minimal human intervention.
In the case of drug discovery, machine learning models comb through the structures of existing compounds with known molecular properties. These models excel at interpolation, meaning they can only be effectively used to predict the properties of molecules that are similar to those in the training set. The predictions become unreliable as the molecules become dissimilar. For example, a model trained to identify cats in photos cannot identify any other animal species.
This is the inherent limitation of AI/machine learning.
Drug hunters often need to extrapolate beyond characterized chemical space to find new molecules. Drug hunters have experimentally characterized far fewer than 200 million drug-like molecules, yet chemical space includes more than 1060 molecules. To put this into perspective, the entire ocean contains about 1047 water molecules and a drop of water contains 1021 molecules. Using machine learning to predict properties of 1060 molecules from a training set with fewer than 200 million molecules risks the equivalent of ‘learning’ there are no fish in the ocean from analyzing one droplet of ocean water!
Molecular training sets are incomplete. And given the vast size of chemical space, it is unrealistic to assume that scientists can generate the data needed to complete the training sets that would be required–it’s an insurmountable challenge.
At Schrödinger, we recognize that developing a completely general approach to predicting molecular properties is not a problem amenable to machine learning. It’s a physics problem.
Physics-based Methods
The other approach for predicting molecular properties is to use rigorous, first-principles methods, also referred to as physics-based methods, that simulate molecular motion at the atomic level. These methods are very powerful in that they can capture all of the complex physics involved in the binding of a molecule to a protein to help predict their molecular properties without the need for existing data.
Predicting how a compound will bind to its target is very complex. For example, the compound and protein will need to adopt a certain shape, the water molecules will have to vacate the binding site, and then the compound will have to bind to the protein. Binding affinity can be predicted using free energy perturbation (FEP) calculations, which fully captures the complexity of the physics of these interactions. Such calculations also require the structure and the small molecule binding site of the protein to be known, which in some cases has not yet been experimentally determined.
However, these calculations are computationally expensive. Given that many desirable molecular properties are anti-correlated, billions of molecules need to be explored to find the most promising drug candidate. There are currently not enough computers in the world to run that many FEP calculations in a reasonable amount of time. This is a limitation of physics-based methods.
Game Changer
AI/machine learning and physics-based methods on their own have limitations that are insurmountable. But when we use them together–by combining the speed of machine learning with the accuracy of physics-based methods–we overcome these limitations. It is by marrying these two approaches that we are advancing the field of drug discovery.
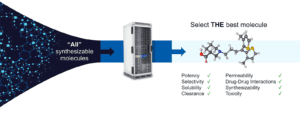
Here is how it works: A computer algorithm builds one billion molecules, for example. We then take a random sampling of only 1,000 of these molecules and compute molecular properties using our free energy perturbation software (FEP+). This step is the equivalent of doing a year’s worth of laboratory work in one day, at a fraction of the cost. We then use the data for those 1,000 molecules to build an approximate machine-learning model that can then be applied to the entire one billion molecules. Using this model, we can score each molecule and take the top 5,000 that best fit our criteria. We then run FEP+ on these 5,000 molecules, which will result in approximately 10 that will be synthesized in the laboratory. These physics-based methods are so accurate that eight out of the 10 molecules will, on average, have the desired molecular property.
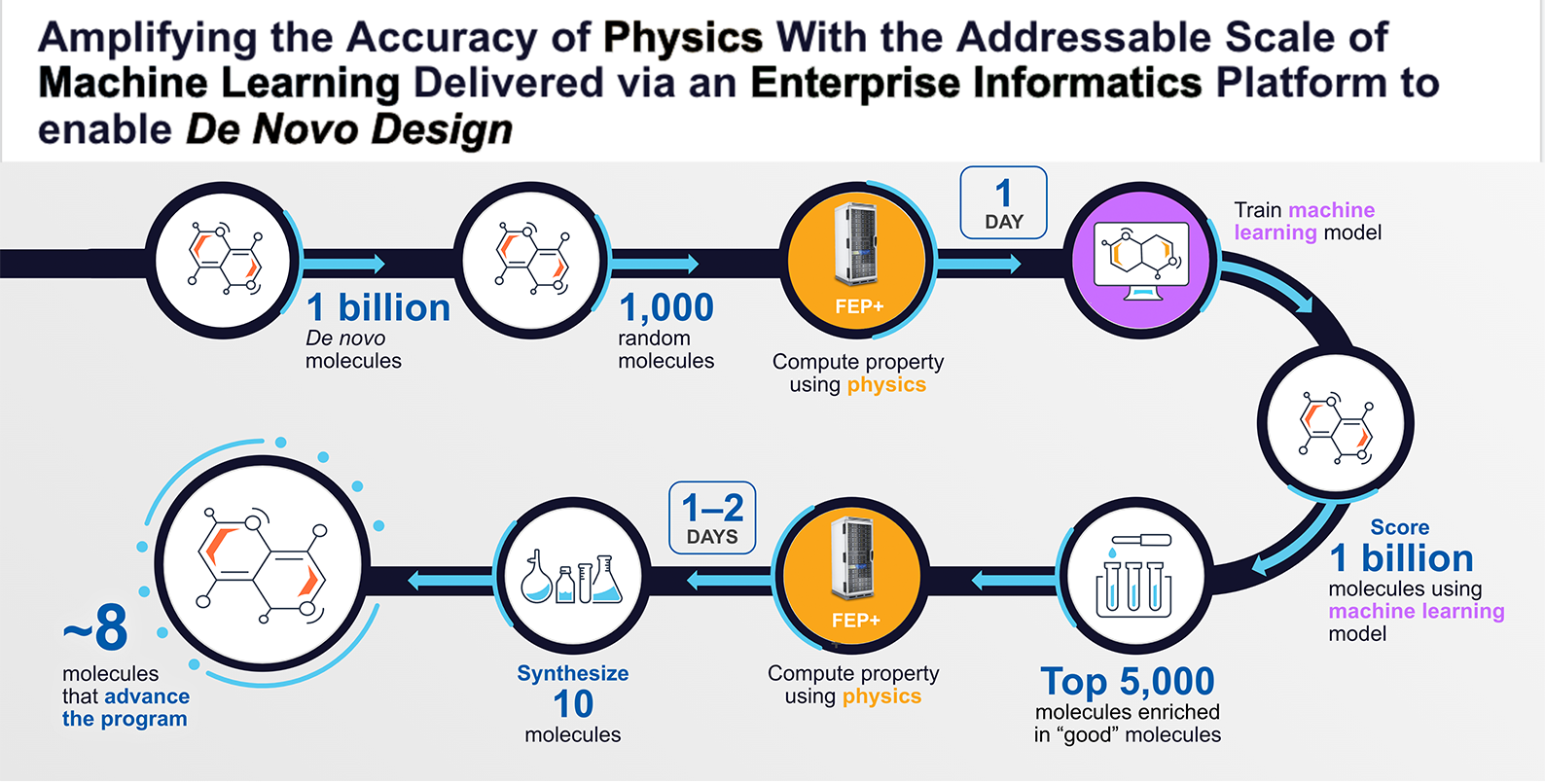
Multiple cycles of this workflow were used to advance our internal MALT1 program. For this program, in total 8.2 billion molecules were prioritized using a physics-based trained machine learning model, approximately 12,000 molecules were analyzed using FEP+, and 78 molecules were synthesized and tested in the lab, one of which became our development candidate that is expected to soon enter clinical development. The identification of the development candidate molecule took only about 10 months.
Where is all this headed? There continues to be exponential growth in the power of computers, the number of proteins for which we know the structure, the number of compounds we can enumerate computationally, and the number of molecular properties that we can accurately predict. Together, these advancements will make the “whack-a-mole” problem a thing of the past.