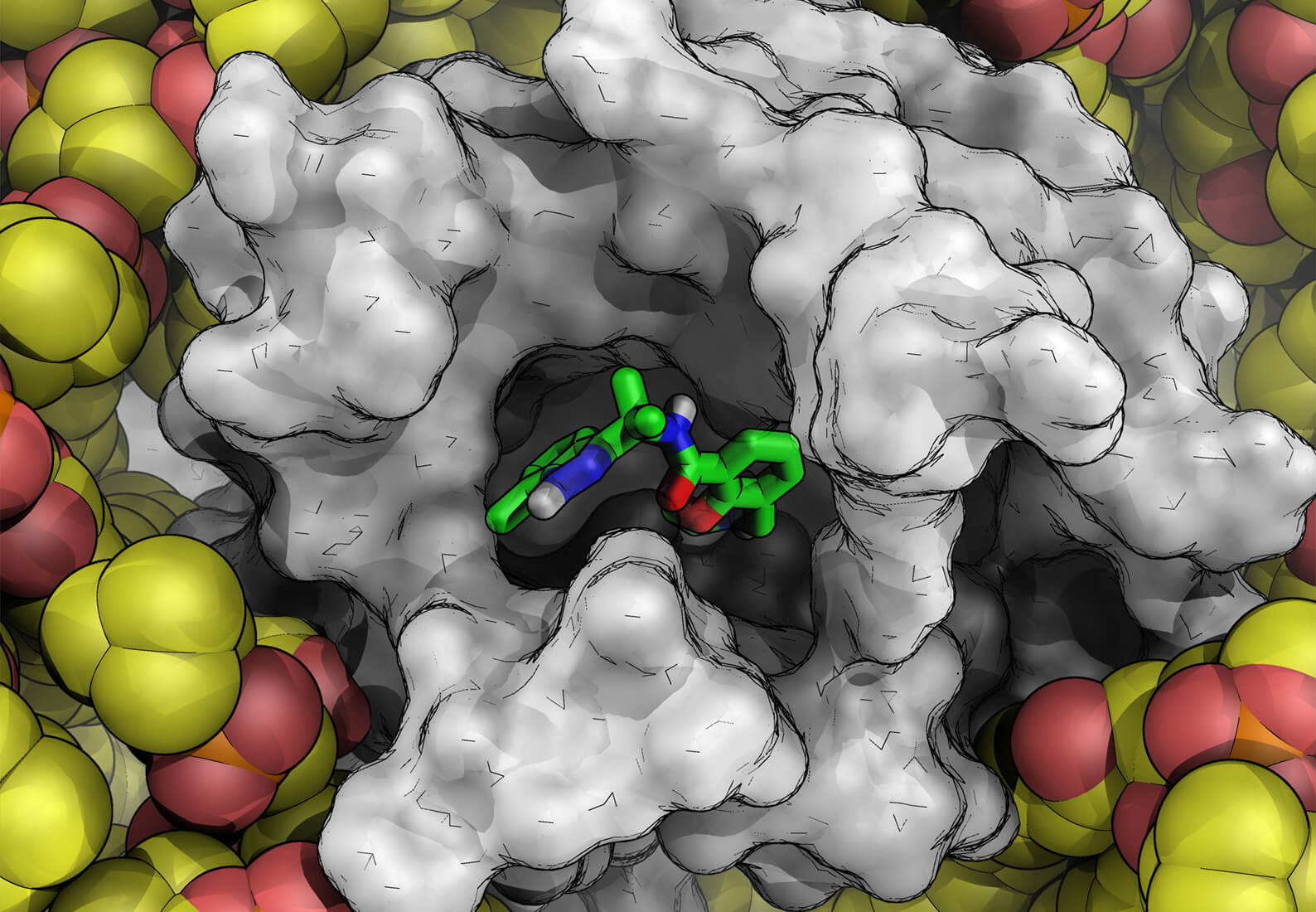





Historically, scientists have only been able to leverage structure-based drug design (SBDD) when high-resolution crystal or cryo-EM structures are available for receptors of interest. This paradigm appeared to be shifting with the release of AlphaFold2, a machine learning-based algorithm purported to be capable of accurately predicting the structure of nearly every protein target in the human genome based on the protein’s genetic sequence. While this breakthrough was celebrated for having the potential to reshape the landscape of drug discovery, questions remained about the quality and utility of the predicted structures.
We recently sat down with Edward Miller, Senior Director of Protein Structure Modeling at Schrödinger, to discuss his experience using AlphaFold models for SBDD.
Let’s dig right in. Can AlphaFold structures serve as a starting point for structure-based design?
Since the release of AlphaFold2, this has become one of the most pressing questions in drug discovery. In our experience, the answer is yes, sometimes, but not without significant computational refinement and validation using methods external to the AlphaFold2 program. We’ve spent the last two years testing such models and putting some of our latest molecular modeling technologies to the task. We’ve found that since AlphaFold2 protein structures don’t include any ligands bound to the protein, which changes the protein’s structure, the predicted structure may contain errors that lead to inaccurate virtual screening results. This same limitation impacts the utility of AlphaFold2 for other activities, such as prospective design of G protein-coupled receptors (GPCRs).
We’ve explored the use of AlphaFold in two main areas: hit discovery and GPCR structure prediction. We’ve published two papers on these topics and have several other research projects in progress.
Can AlphaFold structures be used to find hit compounds?
We conducted a retrospective investigation of this question, and it does appear that AlphaFold structures can provide some utility for hit finding—but with some pretty significant caveats. First, you must know the binding site and binding mode, and second, you must have knowledge of at least one previously identified hit compound to refine the structure, which is not always available.
The first issue is that to be able to perform a structure-based virtual screen of a large number of ligands, the receptor must be treated rigidly, which means even small errors in the structure can impact scoring accuracy and limit the ability to find true hits. We quantified this in our study by performing a virtual screen with AlphaFold structures against 37 targets from a known data set that included proteins with no ligand bound, proteins with ligand bound, and a set of active hits and decoys. Since AlphaFold2 protein structures do not presently include any ligand information, when used out of the box, the models were leading to a lot of active hits getting misclassified as decoys. However, we were able to improve the performance of the AlphaFold model if we docked a known hit molecule using IFD-MD, a molecular dynamics-based induced fit docking technology, which can reorganize the protein to accommodate a binding ligand. Virtual screening calculations using these IFD-MD-improved structures provided more accurate results.
“It is crucial to keep in mind that proteins change their shapes, sometimes quite substantially, when different drug molecules bind to them. As it exists now, AlphaFold2 is unable to model these very important effects.”
Can AlphaFold models be used for structure-based design of GPCRs?
GPCRs are of particular interest to the drug discovery industry at the moment because of the recent explosive success of certain Type 2 diabetes and obesity drugs, which are GPCR-based. However, the complexity and inherent plasticity of GPCR active sites pose unique challenges. Several key hurdles lie in predicting the correct ligand binding mode and establishing robust correlation between computational predictions and experimental data to drive prospective design of GPCRs.
In our study, we wanted to see how AlphaFold would perform on a target the algorithm has never seen. We specifically aimed to select targets of pharmaceutical interest for which neither public experimental structures nor close homologous experimental structures were available—there’s no point in asking AlphaFold for a model of a protein that is 95 percent similar to what is already out there.
To refine the AlphaFold models of GPCRs for prospective design, we employed two physics-based technologies, IFD-MD, to flexibly dock the ligand into the binding site, and FEP+, our free energy perturbation-based in silico binding affinity assay, to quantitatively challenge the model. IFD-MD is necessary because the likelihood that an AlphaFold model is perfect out of the box for any arbitrary ligand series is close to zero. It is extremely common for proteins to be altered in the presence of a ligand (an induced fit); therefore, a single structure almost never encompasses the totality of a receptor’s conformation across all the tight binding chemical matter. This is true of a high resolution crystal structure, and it’s just as true for protein structures from AlphaFold.
After refining the AlphaFold models with our physics-based tools, we were able to show strong correlation between predicted and experimental ligand activity, achieving a level of accuracy comparable to a crystal structure. Our results suggest that AlphaFold models can be used in structure-based design of GPCRs—once they have been properly refined through the application of physics-based technologies.
Are you using AlphaFold and these refinement technologies in Schrödinger’s own discovery programs?
We are using AlphaFold in some interesting ways. In our MALT1 program, a significant structural uncertainty in the binding site posed a challenge in developing an FEP+ model to accurately predict modifications of core and variable regions of molecules. To address this challenge, part of an AlphaFold structure was used to rebuild missing regions and resolve the uncertainty in one of our experimental structures. Ultimately, a large-scale assessment of various FEP+ protocols using machine learning determined that the model with an AlphaFold-supplied loop performed the best. This allowed us to use FEP+ more accurately to predict which new compounds merit the expense of synthesis.
What do these advancements in refining AlphaFold structures mean for the future of drug discovery?
In the limited context of de novo protein folding, the accuracy of the AlphaFold models are truly remarkable compared to the level of accuracy historically obtained with alternative techniques. However, it is crucial to keep in mind that proteins change their shapes, sometimes quite substantially, when different drug molecules bind to them. As it exists now, AlphaFold2 is unable to model these very important effects. Thus, it is important to view an AlphaFold structure as a starting point for a modeling campaign, where given rather small amount of data (20-30 ligands), and knowledge of the binding site, scientists can use computational methods to refine and validate the structures to a level of accuracy necessary to advance a discovery project.
To our knowledge, only the physics-based approaches described here can distill such limited data into a model competitive with experiments and suitable for prospective use. And prospective use of structures generated like these in an active drug program is the final test. We envision this as the future of drug discovery—structure-based drug design using computational structures to predict a molecule’s on target and off-target effects.