A picture is worth a thousand words. How does this long-standing idiom – popularized by early 19th century journalists and advertisers, and attributed to Confucius – apply to drug discovery and compound design? In structural biology, as in many fields, complexity can be simpler to understand when represented graphically. Over 40 years ago one of the most famous molecular structures ever determined was published. The paper in Nature titled “Double Helix at Atomic Resolution” confirmed the existence of “a right-handed antiparallel double helix with Watson-Crick hydrogen bonding between uracil and adenine” described 20 years earlier by the 1963 Nobel Prize in Medicine or Physiology winners, James Watson and Francis Crick.
Single particle cryo-electron microscopy (cryo-EM) has been in use for decades, but a recent “resolution revolution” that allows one to achieve near-atomic resolution has transformed cryo-EM into an increasingly important tool for creating invaluable pictures of protein structures. Cryo-EM allows scientists to lock a protein in its “natural state” by freezing so fast that ice crystals cannot form. Hundreds of thousands of pictures are taken of individual frozen proteins in a myriad of conformations and orientations.
 These images are reconstructed into a three dimensional model that reveals hitherto unknown clues about how the protein interacts with its environment, ligands, and even how it functions. This visual understanding elevates our ability to design molecules that alter protein function.
These images are reconstructed into a three dimensional model that reveals hitherto unknown clues about how the protein interacts with its environment, ligands, and even how it functions. This visual understanding elevates our ability to design molecules that alter protein function.
Growing adoption of cryoEM in academia and industry as well as the formation of collaborative groups like the Schrödinger Cryo-EM Initiative (SCI) signals it’s importance. The 2017 Nobel Prize in Chemistry was awarded jointly to Jacques Dubochet, Joachim Frank and Richard Henderson for their work on the development of cryoEM.
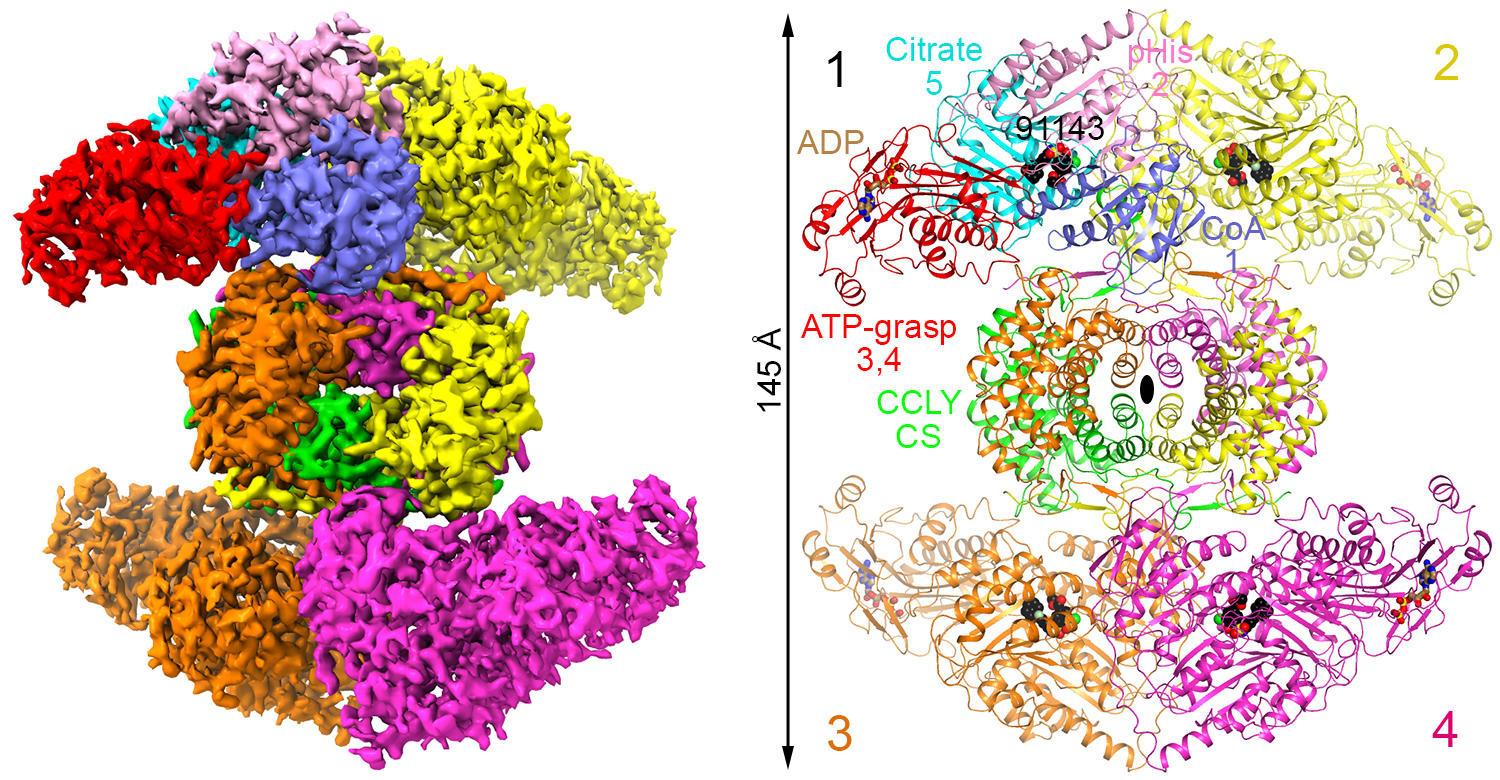
The use of cryo-EM is a key feature of the recent publication in Nature describing the first high-resolution structure of full-length human ATP-citrate lyase (ACLY) pictured below. The game-changing implication of this work is a new generation of discovery programs that benefit from a) protein structure-function insights and binding mode validation enabled by cryo-EM structures and b) structure-based prospective prediction of structure-activity relationships using free energy perturbation (FEP+), a physics-based approach to accurately predict compound potency and many other key compound characteristics.
Understanding the importance of these two methods to discovery programs requires a historical view of drug discovery. The rise of synthetic organic chemistry enabled the identification and large-scale preparation of “non-natural” drug candidates, and was ignited by the ability to prepare analogs of the natural beta-lactam core with a non-natural side chain that had activity against penicillin-resistant bacterial strains. At that time, synthetic chemistry offered the promise that “if a drug could be envisioned, it could be made”. The problem was that we lacked the detailed information at the molecular level about proteins and protein structure-function. Optimizing the activity of known drugs based on preclinical pharmacology models was an empirical but productive approach for many decades.
Engineering innovation (NMR, HPLC, MS) played an important role in spectrometric determination of structures of pharmacologically active compounds at scale and speed for testing in biological assays. Industrial-level drug discovery came from the introduction of small molecule library synthesis and high-throughput screening of millions of molecules aided by robotics and miniaturization of assays. These techniques quickly became a mainstay of the early stages of pharma and biotech discovery programs even though they sample only a fraction of ‘drug-like’ chemical space (estimated at >1 × 10³³ compounds). Concerns over whether there was sufficient chemical diversity in large multi-million compound industrial libraries spawned new approaches for generating hits (e.g. DNA-encoded libraries and fragments screens) as starting points for medicinal chemists.
Along the way, “rational” drug design, also known as computer-aided drug design, emerged, fueled by the significant increases in computer power in the 1980s and advances in protein crystallography. However, being able to accurately predict the interaction strengths of molecules with proteins turned out to be a harder problem than was initially appreciated. In these early days, the pictures didn’t deliver the kind of insights that were needed. Static images of the binding site and molecules weren’t sufficient to enable researchers to predict the interactions of related compounds. Physics-based methods would prove to be the key to unlocking accurate predictions that correlate with assay data.
Let’s now fast forward to the current and future decades of small molecule drug discovery. With access to a growing number of high-resolution x-ray, NMR and cryoEM structures as well as physics-based models to accurately predict compound potency and other parameters (e.g. pKa and solubility) across millions or billions of diverse compounds, we need only synthesize the most promising subset of molecules. Coupling this vast amount of in silico simulation data with machine learning methods that allow teams to maximize and scale the impact of these tools, might lead one to the conclusion that a “picture” — in this case of a detailed protein structure, binding sites and ligand binding modes — is worth a thousand compounds or more. When combined, these innovations are a powerful accelerator in the discovery and optimization of novel, potent, selective inhibitors for biologically and therapeutically relevant proteins.