Arguably one of the most important challenges facing the pharma industry today is the efficient adoption of digital platforms to improve the discovery and development of new medicines. At Bayer, we have tackled this challenge with widespread digitalization efforts across our R&D organization, based on the firm belief that we must evolve in order to excel in the future. While our scientists have been using Schrödinger’s computational software for decades to help discover new drugs, this relationship matured into a strategic technology partnership in 2019 with the formation of a large-scale collaboration between the two companies. The result is a drug design platform that we’ve termed at Bayer the NovaDesign platform.
NovaDesign is a technology platform that combines Schrödinger’s and Bayer’s best approaches to intelligently create and filter through tens of millions of virtual compounds to find completely novel lead structures as the basis for future therapeutic candidates. It does so by leveraging Schrödinger’s technology, which encompasses rigorous physics-based methods, machine learning, and a centralized ideation platform to capture data and collaborate across teams, and in silico models to predict properties such as compound absorption, distribution, metabolism, and toxicity (ADMET) and chemical synthesizability based on Bayer’s vast and collective knowledge and data.
Ultimately, Bayer plans to use this collaborative platform to rapidly design large numbers of synthesizable virtual molecules and predict their properties across many discovery programs in our pipeline. We’re very excited by the progress being made just one year into this collaborative effort, as the platform has already impacted several active projects at Bayer.
With the right digital partner and the right investments, pharma companies can ensure they stay on top when it comes to successful drug discovery, to continue to help patients.
How it works
The process starts with library creation, combining a collection of commercially available reagents, Bayer’s reagent library, and reaction-based enumeration. This is followed by a robust filtering process, including filtering for basic phys-chem properties, synthesizability, and amenability to the advanced modeling applied towards the end of the workflow.
We then apply machine learning based filtering with the goal of generating about 10 million compounds by the time we reach the docking step. Following docking, we upload the top 100,000 compounds into LiveDesign, Schrödinger’s centralized enterprise informatics platform that facilitates data aggregation, model deployment, and team collaboration. This is followed by an active learning free energy perturbation (FEP) approach, which results in the selection of top-scoring compounds. FEP uses extensive molecular dynamics calculations on a high-performance computing infrastructure to simulate most realistically the binding of small molecules to target proteins. The iterative loop of physics-based protein-ligand affinity prediction on a subset of compounds, training of fast machine learning models on this data, and selecting the next round of compounds for physics-based affinity prediction based on these models ultimately yields a low number of virtual compounds (~10-100) that are then processed with even more accurate, full-cycle-closure FEP calculations. This results in a few highly computer optimized compounds for visual inspection and synthesis prioritization. In short, the active learning approach allows us to estimate potency for a large number of virtual compounds rapidly.
We then review the remaining compounds in a team of medicinal and computational chemists to identify the best ones to synthesize in the lab. Once we move on to in-vitro testing, we try to pinpoint the very best compounds: the “winners,” if you will.
And it works.
Early successes
NovaDesign is being applied across many active discovery projects. In one of our very first runs in an early lead-finding project, we enumerated a million compounds and ultimately the team selected one highly promising compound for synthesis; this compound was only 25 percent “new” compared to the starting compound but showed improved potency while having a comparable beneficial physicochemical profile.
In another notable project, the workflow identified molecules that are 75 percent “new” compared to the starting compound, with an enhanced combination of properties. The impact of the platform is astounding in its ability to uncover brand-new active substances that encompass highly important properties. In this project, we just synthesized around 20 compounds to deliver a molecule with high novelty and quality that usually would qualify as a starting point for a lead optimization project.
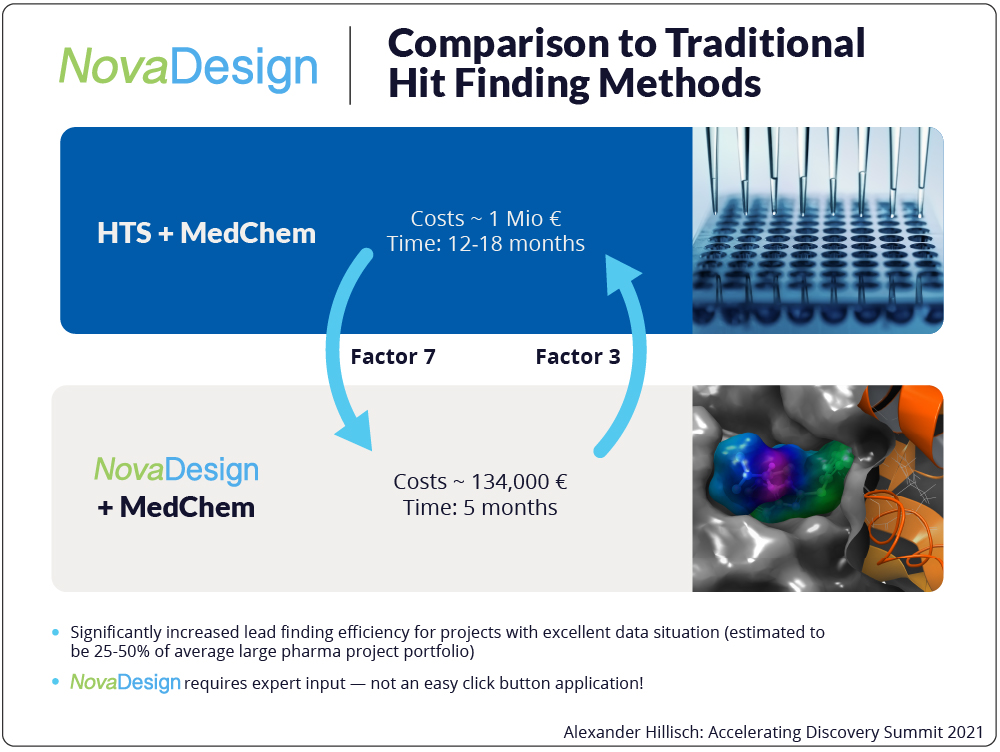
This holistic de novo approach is a game-changer, enabling Bayer to gain efficiencies and ultimately discover drugs faster. It also opens the door to more possibilities in drug discovery and higher quality molecules – molecules that may have otherwise never been discovered with tried-and-true experimental efforts. The sheer speed and efficiency resulting from NovaDesign also positively impacts cost, making possible the discovery of molecules with far less expense per project since we are synthesizing and testing only the highest quality lead molecules. It has to be mentioned that this approach is not applicable to each and every drug discovery project. The existence of a high-quality protein complex structure and some robust assay data to achieve a predictive FEP-system renders NovaDesign applicable to about a quarter of the small molecule drug discovery projects overall.
Apart from the immediate benefits, this type of innovation further supports a growing trend in the drug discovery space in which protein target structure information (X-ray, Cryo-electron microscopy, and/or AlphaFold) will only become better and more accessible to drug hunters over time, provided they are starting with good data. Additionally, it underscores the importance of data-sharing among pharma companies in general, opening doors to new data-generation methods. With the right digital partner and the right investments, pharma companies can ensure they stay on top when it comes to successful drug discovery, to continue to help patients. Our collaboration with Schrödinger, and the early success we have seen so far, as a result, is a testament to that.